
by Kimberly Allen R.N.
Huntington’s Disease also called Huntington’s Chorea is an inherited disorder that affects the nerve cells in the brain. Unlike in many other inherited disorders Huntington’s disease is caused by an autosomal ‘dominant’ gene mutation. This means that instead of needing 2 mutated genes you only need 1 mutated gene to develop Huntington’s disease. Huntington’s disease is more prevalent in people of Western European descent.
It is estimated by the Huntington’s Society of America that 1 out of every 10,000 Americans has the disease as well as 150,000 Americans that have a 50% chance of developing the disease. However some experts believe the numbers are actually much higher because the disease usually doesn’t manifest until mid adulthood there are many that not on have the disease but are unaware of it yet, there are many of those that have had children that have inherited the gene mutation and are unaware of it. It affects both men and women equally and Caucasians are 70-100 times
more likely to develop Huntington’s disease than those of Asian or African descent.
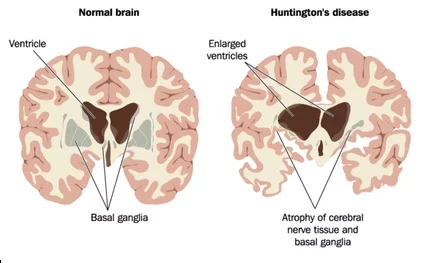
Though the exact mechanism of action is unknown, it is known that the Huntington’s gene that is mutated causes a mutated form of the protein “huntintin”. This mutation gradually causes damage to certain cells in the brain. These cells affect muscle coordination, cognitive function and behavior.
The symptoms of Huntington’s disease usually present when a person is between 40 – 50 years of age, however they can develop earlier or later. Some have what’s called juvenile Huntington’s disease, they develop symptoms early usually before the age of 20. Most experts say that the earlier the onset of symptoms the faster the disease progresses. Early symptoms of Huntington’s disease varies significantly from person to person. In some people their family may notice mood swings and changes in behavior like being more irritable, depressed, apathetic or angry. Some people may have hostile outbursts or suffer bouts of deep depression. In many people the disease firsts manifests with uncontrolled movements in the fingers, face, trunk, and feet that tend to increase with stress. As the disease progresses it causes difficulty with walking and can affect swallowing, eating and
speech.
There is no cure for Huntington’s disease, however there are medications that can help control the symptoms including the
involuntary movements and psychiatric disorders. There is also a new medication available called tetrabenazine that is the first medication approved for treating Huntington’s disease in the US.
It’s important to remember that many of the medications used to treat the symptoms of Huntington’s disease have side effects that are similar to those of the disease so it can be difficult to determine if a symptom is part of the disease or a side effect of medication. One of the most important components of any treatment plan for Huntington’s disease is ‘exercise’. Those that maintain their physical fitness by including exercise in their daily routine do better than those that don’t.
Nutrition is also a crusial part of caring for Huntington’s disease. As medical science and treatments for Huntington’s disease has advanced so has the quality and length of life after the onset of symptoms.
References
-
NINDS. “Huntington’s disease (overview).” Updated Apr 8, 2025. NINDS
-
GeneReviews®. “Huntington Disease.” Updated resource. NCBI
-
MedlinePlus Genetics. “.” Updated Apr 16, 2025. medlineplus.gov
-
The Lancet Neurology. “Potential disease-modifying therapies for .” 2022. pubmed.ncbi.nlm.nih.gov
-
The Lancet Neurology. “A biological classification of.” 2022. thelancet.com
-
Nature Genetics. “Cell-type-specific CAG repeat expansions and toxicity…” 2024. nature.com
-
HDSA. “Overview of .” hdsa.org
-
MedlinePlus Genetics. “-like (HDL) conditions.” Updated Jun 28, 2024. medlineplus.gov
-
NINDS. “: Hope Through Research” (PDF). May 2025. NINDS



{kind=link}